04 June 2019
Newly published multi-laboratory study provides utility and validation of the use of ctDNA reference standards
Share:A recent study was published, sharing findings on circulating tumor DNA reference standards. Below, Russell Garlick, Chief Scientific Officer at SeraCare, shares his thoughts on the new study.
I am pleased to share findings from a newly published peer-reviewed study with foundational circulating tumor DNA (ctDNA) pre-analytical and analytical testing in multiple technologies and assay chemistries. The study, “Multi-laboratory Assessment of a New Reference Material for Quality Assurance of Cell-Free Tumor DNA Measurements,” was just published in The Journal of Molecular Diagnostics (He, Stein et al. 2019). I use the term “foundational” since validation of the technology used to design, develop, and manufacture the ctDNA reference materials that are central to the study helps to establish a critical tool set that is required to standardize testing through comparison analysis, providing a much needed confidence boost to liquid biopsy-based cancer diagnostic or disease monitoring tests.
Before I discuss the study, I’ll highlight two recent publications that demonstrate why we need high-quality validated reference standards manufactured in large quantities, and why ctDNA testing is so promising for cancer patients.
The Promise of Liquid Biopsy
The promise of liquid biopsy cancer diagnostics is to use a minimally invasive procedure to detect metastasis or disease recurrence as early as possible. A hallmark for cancer treatment is that early detection and targeted treatment often leads to longer disease-free periods and better outcomes. ctDNA driver mutations or resistance markers are shed from the tumor into the circulation and can be detected in plasma. Many studies have shown concordance between driver genes found in the primary tumor to those found in blood. Sensitivity has been an issue since measuring just one variant allele mutation, e.g. a single nucleotide variant C>T mutation, for every thousand wild-type reads is a technical challenge when the sample is a blood draw.

Widespread adoption of a technically complex assay like ctDNA is even more complicated since it is offered by dozens of testing labs as an LDT and analytical performance is kept confidential making comparisons from lab to lab or assay to assay very difficult. To address this issue, AstraZeneca took matters into their own hands and initiated a comparison study of their own. This was just published in the Journal of Clinical Oncology in March. The study (Stetson, Ahmed et al. 2019) used patient samples to assess concordance between four labs offering ctDNA testing.
Not unexpectedly, discordance was especially evident at the very lowest levels <1% VAF. In the Stetson study, twenty-four matched tumor plasma samples were sent to four commercial labs. Variant-level concordance was high in plasma samples above 1% variant allelic fraction (100% PPV for 3 of the 4 labs) but very poor overall (PPV ranged from 36% to 80%). As will be discussed below, ctDNA assays must approach 0.1% VAF to meet early cancer detection needs of patients.
These results not only made clinicians pause when ordering tests, they also negatively impacted pharmaceutical companies validating genotypic biomarkers or enrolling patients in trials. The discordant results also prompted several projects to improve ctDNA testing via inter-laboratory concordance studies. One well-designed study underway is the Foundation for NIH Biomarkers study. This effort seeks to validate commercially available reference materials as aids to the development, validation, and QC NGS analysis of ctDNA-based molecular assays. The first phase (1a) of the F-NIH study is NGS assay analytical validation and a 1b study with patient samples is designed to assess commutability of the reference materials. The second phase is clinical validation of up to 14 genotypic biomarkers. BloodPAC is also engaged in inter-laboratory concordance studies for the same purpose.
On the positive side of ctDNA testing, recent reports from Memorial Sloan Kettering Cancer Center (MSKCC), Dana Faber Cancer Center, and several other cancer treatment hospitals/laboratories are very encouraging and highlights the potential of ctDNA testing in cancer patient management. Li, et al. show that ultra-deep sequencing (20,000 X) with clonal hematopoiesis filtering, can detect de novo driver mutations at <0.5% VAF (Li, Janku et al. 2019). Here is Figure 1c. showing lung cancer driver mutations and resistance mutations for 7 genes.

Note the overall number of driver mutations, especially those below 0.5% (green arrow) and the number of resistance mutations in red. Actionable resistance mutations including EGFR T790M, EGFR C797S, and ERBB2 amplification were found frequently enough to raise expectations that one of the main purposes for blood testing, early resistance monitoring, can be met. In addition to resistance, the authors state that their method is quite promising since targetable lung cancer driver mutations could be identified in plasma prospectively when tissue failed or could not be biopsied due to patient safety concerns.
Validation of ctDNA Reference Material Manufacturing Technology
Now let’s get back to the aforementioned multi-laboratory assessment. No need to repeat everything in this blog post that is reported in the manuscript, but I do want to provide contextual information to those readers who are interested in liquid biopsy standardization, or who are simply just interested in why we did the work in the first place. Let me highlight three observations from the report:
- An assessment of sample prep or A.K.A. the pre-analytical phase. I mentioned earlier how difficult it is to measure a single base change from the tumor cells from a blood draw. A prerequisite is achieving a high and consistent DNA extraction yield. Reference material stability is also critical considering manufacture and distribution around the globe. The data in Table 2 (He et al.) shows that the reference materials at 0%, 0.125%, 0.5% and 2% VAF manufactured by this novel technology can be used to compare DNA yields from 7 commercial DNA extraction methods with statistical precision. For example, one extraction kit using a manual extraction protocol will yield more DNA than the automated protocol (P< 0.05) for the same technology. One lab also shows that by adding RNA to the DNA elution buffer, the DNA yield almost doubles demonstrating the carrier effect works yet again. Maximizing DNA extraction is the first critical step to achieving maximal variant detection sensitivity. To accomplish this aspect using patient samples would have required about a unit of blood from a cancer patient, thus not a feasible approach.
- The use of droplet digital PCR to establish a reference value or “ground truth” for the material to which other methods can be compared. We show ddPCR is a reliable, low cost, and accurate method to characterize multiple types of variants (40 in total) to very low levels 0.125% VAF (~19 copies / ml). ddPCR was used to establish the initial “ground truth” variant allelic fraction measurements for comparison to low plex single gene assays and to high plex multigene NGS assays. At SeraCare, each variant in the reference material is quantitated using a validated ddPCR assay.
- The results provide a preliminary view of analytical sensitivity to 0.125% VAF for multiple variants SNVs, insertions and deletions. As discussed above, ctDNA assays must approach 0.1 VAF% to be sensitive enough for routine use. One of the most promising approaches to high sensitivity testing is the SimSen-Seq Assay developed by Drs. Tony Godfrey and Anders Stahlberg. Shown in Figure S2 from (He et al.) is the SimSen-Seq NGS Data for Reference Materials for Four Variants: PIK3CA (H1047R), KRAS (G12D), BRAF (V600E), and NRAS (Q61R). The errors bars are 1 standard deviation from n = 3 measurements and clearly show reproducible detection from 2% to 0.13%.

Lastly and not to be overlooked is overall variant detection. Figure 6 compares all NGS results tested by each of the seven laboratories at n=3 and shows detection precision at these levels.
Reference Materials are Necessary for Clinical Success
We are in the earliest days of ctDNA adoptions into clinical practice, thus the ability to compare assay results using a reference material made in bulk by GMP procedures is critical not only to assess the utility of the technology used to make the reference materials, but also to establish ground truth measurements that assay developers and clinical labs can rely on to standardize and make improvements. This technical approach to design, develop, and manufacture a set reference material for IVD manufacturers and clinical labs to identify and correct assay biases before expensive clinical trials are started is fundamental to the advancement of liquid biopsy.
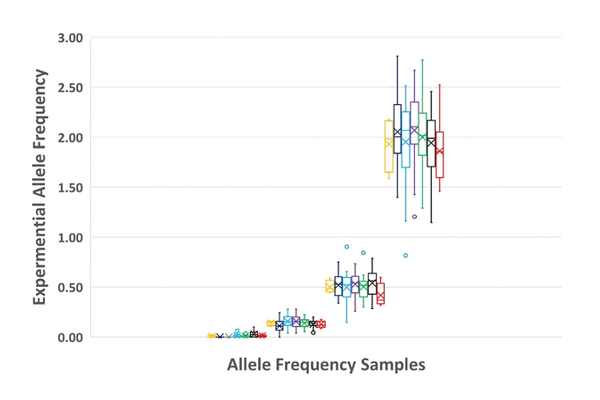
Figure 6 compares all NGS results tested by each of the seven laboratories at n=3 and shows detection precision at these levelsSo how did the collaborative effort start? In 2016, SeraCare and The National Institutes of Standards and Technology (NIST) jointly funded a Collaborative Research and Development Agreement (CRADA) to establish the requirements for the most useful reference materials and controls for ctDNA measurements. This is a collaboration of scientists and our Gaithersburg neighbors with an aligned purpose. SeraCare is focused on the design, development, manufacture, and distribution of quality control materials applied to clinical genomics, and NIST’s mission is to promote innovation and measurement science. NIST’s core competencies include the development and use of standards for biological applications including liquid biopsy. NIST directs resources to areas of greatest commercial need, and clinical genomics is a high priority. The NIST lab is very experienced with complex assay quantitation, statistics, qPCR, and ddPCR assay development and validation. Within the CRADA umbrella, we set out to understand what the requirements are for a fit-for-purpose reference material and to assess the variables of a liquid biopsy workflow through an inter-lab study. NIST was highly instrumental in recruiting leading laboratories for interlab testing including Frederick National Laboratory for Cancer Research, Boston University School of Medicine / Sahlgrenska Cancer Center, Johns Hopkins University School of Medicine, University of North Carolina School of Medicine, Asuragen, ArcherDX, and National Cancer Institute.
The design requirements for a ctDNA multiplex reference material are complex and many, and earlier in the blog I stated the results are foundational for future products. By this I mean we now have a very good assessment of how these materials work across a range of assay formats and extraction kits.
Let me highlight some important lessons learned for the performance of the reference materials from the study. First at the molecular level. The DNA must have similar size 160-170 base pairs to natural ctDNA, and it did. The mix should contain clinically relevant variants that are difficult to detect such as EGFR exon 19 deletions c.2236_2250del15 and be able to detect more common, easier to detect SNVs BRAF c.1799T>A. The mutation mix used for the study contained 40 variants (SNVs, insertions, and deletions) and it demonstrates we can make a highly multiplexed material that tested just once in an NGS assay, generates a tremendous amount of high quality data, easily increases productivity, and saves the lab precious sequencing costs by not having to run multiple samples at $400 per sample.
Through many designs of experiments at SeraCare R&D, we’ve optimized design of the biosynthetic variants, spike-in to genomic DNA, fragmentation, sizing, and end-repair to best represent natural patient-like reference standard. The %VAF must be manufactured to be below a clinical cutoff. Based on the (Li, Janku) study discussed above, ~7% patients have driver or resistance mutations between 0.1-0.5% VAF. From a manufacturing viewpoint, this means, using BRAF V600E as an example, that for every 1000 molecules there are 999 “T’s” and just 1 of the variant nucleotide sequence “A”; see COSMIC 476 c.1799T>A. We applied precise and accurate manufacturing approaches and QC measurements afforded by allele-specific ddPCR for each variant in the mix at these lowest target levels 0.125% VAF. These same methods can be applied to ten-fold less.
Not to be overlooked is the macroscale, the physical form of the reference material, and how the DNA is packaged and in what diluent. Although the half-life of natural ccfDNA in normal circulation is very short, ~16 min due to tissue uptake and degradation, it has been shown that naturally circulating DNA is mostly protected from nucleases (Lo, Zhang et al. 1999). At SeraCare, we decided to replicate this feature of ccfDNA by mimicking the apoptotic body form found in circulation by using a highly reproducible, liposome encapsulation method developed and patent pending by SeraCare. This method protects the DNA from nucleases and damaging polymerization, and it improves long term stability at 4oC. Finally, the reference material was formulated in a synthetic SeraCon Matribase (SeraCare). This diluent is formulated to ensure total protein of 5.9-6.6 g/dL (human albumin); endogenous ions, salts, glucose etc. are always at the same physiological specification, and it ensures there is no assay-interfering hDNA carried over from human plasma or enzyme inhibitors that might impact library prep or sequencing. The data also show that the material for this version is not ideally suited for specificity assessment due to background noise in the wild type material. This feature has already been addressed in newer versions of the technology which are more suited for specificity testing. (Stay tuned as this is a future blog post topic).
Conclusions
So, based on the data, where do we stand and where do we go from here?
- Flexible design. We have a flexible reference material manufacturing technology. Each of the steps described above to make reference materials with different mutations and at differing allelic fractions is easily reproduced for different assay formats and cancers.
- Large scale. The materials are made in large scale for broad distribution to clinical labs around the world. The materials can be used for proficiency testing or for in-kit positive controls.
- Stable. The materials are stable in SeraCon Matribase for > 3-year real time at 4oC.
- Analytical sensitivity. Due to the design features, materials can be made to bracket clinically relevant cut-offs and help set assay sensitivity levels to 0.125% VAF or lower.
- Routine QC. Guidelines like New York State Department of Health and the FDA recommend daily run controls and the form and function of these materials are ideal to QC systems and daily QC.
- Data to make improvement for the next-generation product. The results identify the need for an improved method to assess specificity. We have solutions that are being tested and implemented in new product designs. Stay tuned.
This blog post was originally published on the SeraCare blog, Genomic Precision. Read more background on this blog post here.